After exploring real-world data sources across the Nordic countries, we now pivot to how this data is used in regulatory evidence generation.
One important example is Post-Authorisation safety Studies (PASS).
The PASS framework is specific to the European Union regulatory system.
Other regulators, such as the FDA, generally refer to these as post-approval studies.
PASS are conducted after a medicine is approved to further identify, characterise, or quantify safety risks.
Most PASS are non-interventional, relying on real-world data from registries, electronic health records, and observational datasets.
Some PASS are conducted voluntarily by marketing authorisation holders.
Others are requested by regulators as part of post-authorisation obligations.
Regulatorily requested PASS generally fall into three categories:
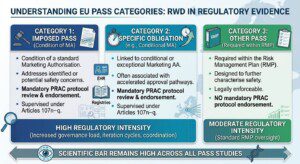
Category 1: Imposed PASS
➡️ Condition of a standard Marketing Authorisation
➡️ Addresses identified or potential safety concerns
➡️ Mandatory PRAC protocol review and endorsement
➡️ Supervised under Articles 107n–q
Category 2: Specific Obligation
▶️ Linked to conditional or exceptional Marketing Authorisation
▶️ Often associated with accelerated approval pathways
▶️ Mandatory PRAC protocol review and endorsement
▶️ Supervised under Articles 107n–q
Category 3: Other PASS
➡️ Required within the Risk Management Plan (RMP)
➡️ Designed to further characterise safety
➡️ Legally enforceable
➡️ No mandatory PRAC protocol endorsement under Articles 107n–q
In practice, Categories 1 and 2 are typically the most resource-intensive.
The driver is not necessarily scientific complexity.
It is regulatory intensity.
Mandatory PRAC protocol review increases governance load, iteration cycles, and coordination requirements.
From a scientific standpoint, the bar should remain high across all PASS studies.
What changes is the regulatory scrutiny and the operational margin for error.
Which PASS category has posed the greatest operational challenge in your experience⁉️